Finally getting round to explaining what set off my last rant about MPS. Of course, I always hate MPS (who wouldn’t when your child has been diagnosed with a life-limiting illness?), but I found last week’s hospital trip particularly hard.
So here it is – the good, the bad, and the ugly. Though as I always prefer to end on a positive note if I can, it’s actually the ugly, the bad and the good!
The Ugly
As you may have read before, the clinical trial Pudding is on had disappointing first year results. Before the boys received their doses this time, our consultant (who also runs this phase of the trial in the UK) gathered us parents together to explain what he has heard, and answer any of our questions. He wasn’t able to give us too much information as the full results are embargoed until February when they will be announced at a conference. But what he could tell us was that he was more heartened by the results than he had expected.
The reason I’m still calling it the Ugly is that analysing data for such a small group is …well… complicated. Without going into a whole essay about the mechanics of designing clinical trials (I find it fascinating, but you probably wouldn’t!) one year of data is just not enough to show clear benefits. So their next step is possibly to include data from other studies done previously which show the normal course of decline in MPSII. Not a straightforward process, but there is potential.
Of course, there will still be the issue of getting agreement from NICE and NHS England to fund it if the drug is approved. But I’m trying to hold onto something our doctor also said about the many battles he has had to fight in his clinical career. ‘I’ve realised that the only way I can get through, is by dealing with them one step at a time.’
The Bad
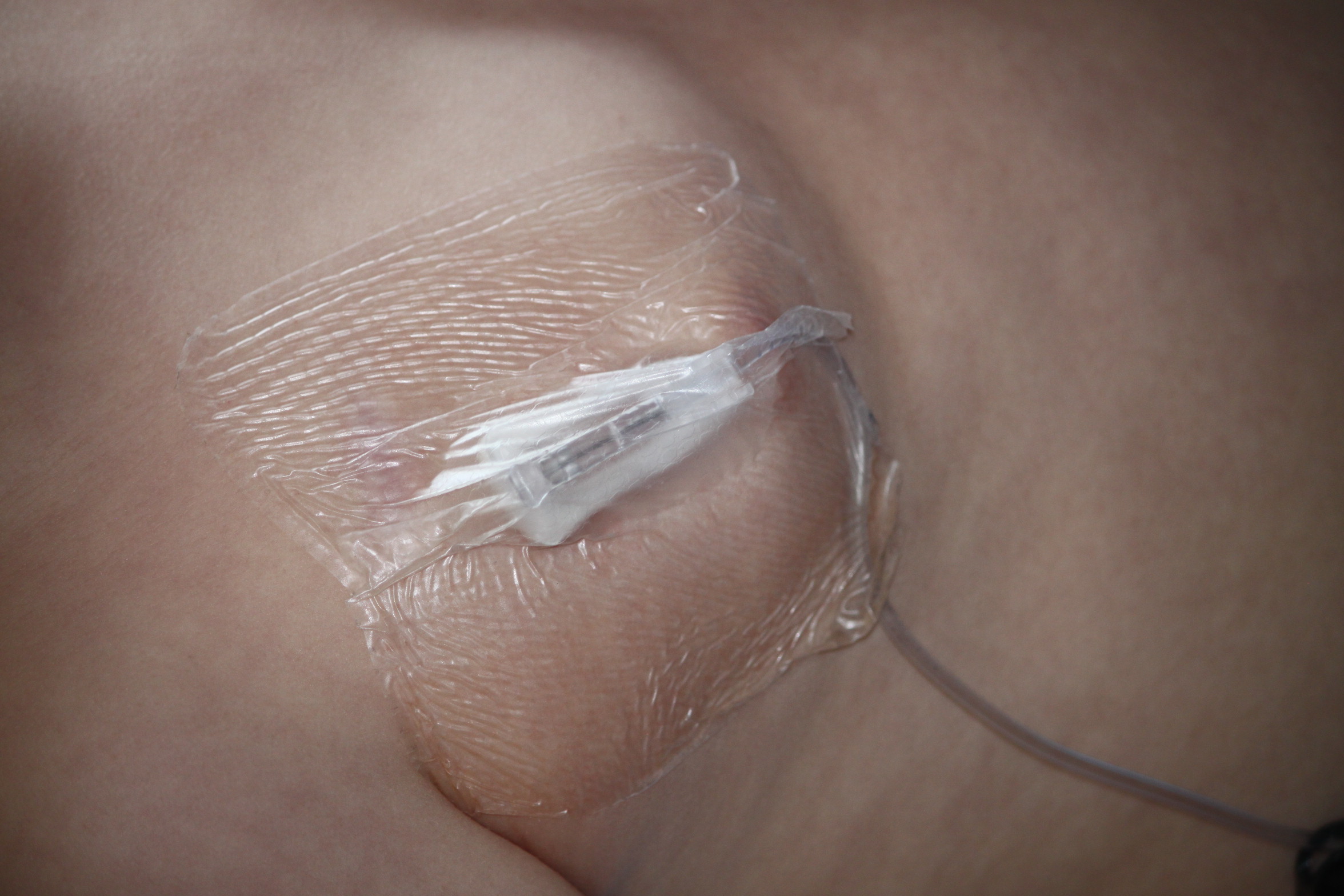
This is the one that knocked me for six. After a bad night’s sleep on the ward (Pudding was still climbing out of bed and switching the lights on and off until nearly 11pm) and the morning’s discussion on trial issues, I had another talk with the consultant. He told me that Pudding has developed antibodies to the enzyme infusion that he receives every week.
Again without going into all the details (lesson on cell biology, anyone?), the basics are that all sorts of different antibodies circulate in the blood. The ones that we really don’t want to see are neutralising antibodies which stop the enzyme being taken up into the cells to do their job. And yes, those are the ones that Pudding has.
These results are actually a year old, so there is a possibility that more recent results will show that the antibodies have gone down again. It’s unlikely though, as there have been a few other reasons to think that the enzyme is just not working as well as it should be for him. Of course, without the enzyme clearing away as many of the waste sugars, they will be building up again, and potentially causing new damage to his organs, joints and so on. So…next stage will be to think about ways to get round it. This will probably mean some form of immune suppression drugs.
The news wasn’t entirely unexpected. Some boys with this condition have a small ‘spelling mistake’ on the DNA, meaning that their body produces a faulty version of the enzyme or just not enough of it. Pudding, however, has a full gene deletion. So the synthetic enzyme he gets is completely foreign to his body, and hence…antibodies.
In the grand scheme of things it’s not the worst news in the world. But it certainly wasn’t what I wanted to hear.
The Good
Yes, that’s it from the depressing side! Yay!
Even in the depths of this horrible MPS world, the silver lining is always the other people that support us along the way. Our lovely doctor, who cares so much for each and every one of his patients and hates giving us bad news. The nurses and play specialist who look after Pudding so I can off by myself for a cry. And of course, my fabulous, wonderful MPS family. This hospital visit was the first time in ages that all four boys on this phase were treated on the same day, so I could have a chat with the other parents.
When I got our bad news, one of them gave me a massive hug with a tear in his eye. Hugs that come from someone who truly understands what you’re going through are the absolute best. They can never make things completely better, but it’s a bloody good substitute!
PS. We do have another bit of good news that I’ve heard this week, but I won’t write about it until we’ve got the official letter!




 For us, the main issue has been Pudding’s health. Not his health now (anyone who sees our photos and videos knows that despite his life-limiting diagnosis he still remains ridiculously healthy) but what is to come. We’ve known since February last year that his body was producing antibodies against the enzyme replacement treatment. Yes, antibodies against that very treatment that is meant to be keeping him alive.
For us, the main issue has been Pudding’s health. Not his health now (anyone who sees our photos and videos knows that despite his life-limiting diagnosis he still remains ridiculously healthy) but what is to come. We’ve known since February last year that his body was producing antibodies against the enzyme replacement treatment. Yes, antibodies against that very treatment that is meant to be keeping him alive.