Another Friday, another infusion.
A few people lately have been aski ng about Pudding’s treatment, so I decided it’s time for a blog post about it! Hunter Syndrome means that he is missing a particular enzyme that goes by the snappy name of iduronate-2-sulfatase. The enzyme would normally clear away waste sugars once they have been used by the body. Without it, the waste sugars build up and cause all sorts of problems. So every week, Pudding gets an infusion of synthetic enzyme to remove the waste and keep things working. This is called enzyme replacement therapy or ERT.
ng about Pudding’s treatment, so I decided it’s time for a blog post about it! Hunter Syndrome means that he is missing a particular enzyme that goes by the snappy name of iduronate-2-sulfatase. The enzyme would normally clear away waste sugars once they have been used by the body. Without it, the waste sugars build up and cause all sorts of problems. So every week, Pudding gets an infusion of synthetic enzyme to remove the waste and keep things working. This is called enzyme replacement therapy or ERT.
For the first few months we had to travel to Manchester for this every week, but now life is much easier with treatments at home.
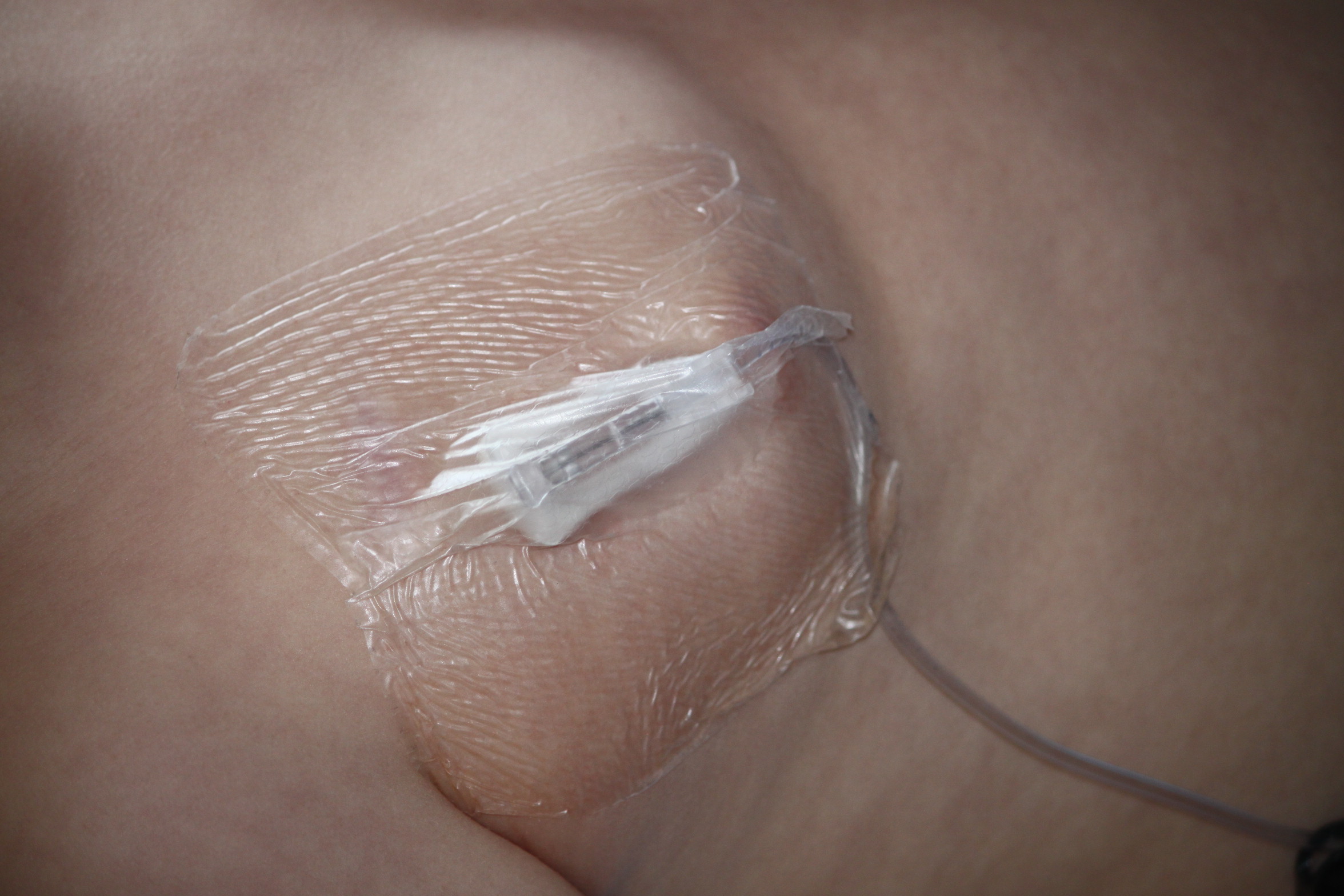
8.15am – I put emla cream on the site of Pudding’s port. This is a device just under the skin of his chest that then feeds his infusion directly into one of the veins going to his heart. The cream means that it will be numb in time for the needle later.
8.30 – We drop T at school. Pudding objects because he wants to go in too. He doesn’t understand why Fridays are different.
9am – Our nurse arrives. Once Pudding has rushed to the door and said hello, he starts signing and saying ‘TV’. It’s good that he associates her with nice things, despite everything! She checks his temperature and gives him some pre-meds (paracetamol and citirizine) to stop any reactions to the treatment. He’s not keen on having medicine, so we have to catch him before he can run away.
9.30am – The nurse gets a sterile tray prepared and is ready to access Pudding’s port. I sit on the floor with him in front of me and wrap all my limbs around him to pin him down. As long as the TV is on he’s pretty good these days – gone are the times when we needed an extra person to help hold him. The nurse uses a special gripper needle to pierce his skin and go into the port.  It is taped down so that it can’t move during treatment and has a thin tube attached to it that his medicine goes through. Before I can release him we also have to try and get a blood pressure reading. Sometimes we’re lucky and get it first go, sometimes it takes several tries before he stays still enough.
It is taped down so that it can’t move during treatment and has a thin tube attached to it that his medicine goes through. Before I can release him we also have to try and get a blood pressure reading. Sometimes we’re lucky and get it first go, sometimes it takes several tries before he stays still enough.
10am – We get the highly expensive, magic enzyme out of the medical fridge. The nurse adds it to a bag of saline so that it can be infused slowly into Pudding’s port. (If it was added in all at once it might cause reactions, and also would be more difficult for the body to absorb.) We have a small electric pump that pushes the enzyme and saline mix through the line at a set rate. When we first started home treatments this was held in a shoulder bag which Pudding refused to wear; we had to hover near him for the whole treatment ready to pick it up and follow him whenever he moved. Not ideal! Now we use a little rucksack – like most aspects of treatment, Pudding is not keen on us connecting the line and putting the rucksack on, but once started it doesn’t seem to bother him.
For the next few hours, Pudding is free to watch TV, play, go outside or whatever he likes within reason. We do have to be careful that the needle doesn’t get bashed as that might mean having to access again – the pump will beep to let us know if there’s a blockage in the line and the nurse checks it every so often.
Noon – A dose of ibuprofen.
1pm – More citirizine. By this dose he tends to give in quite easily and opens his mouth for it even without being asked.
1.50pm – The pump beeps to let us know that the saline bag has finished. The nurse puts on another bag to flush any enzyme that is still left in the line.
2.10pm – The flush is finished and we can disconnect the line and take off his rucksack. I have to get back into position on the floor for de-access. First there is an injection of heparin into the port to prevent any blood clots inside it before the next infusion. Then it’s time to remove the needle. Probably his least favourite part is having the dressing peeled off. More paracetamol. Another blood pressure. And we’re finally done. The nurse finishes off all the paperwork, and Pudding ‘signs’ it off on the tablet.
Just in time to head off on school run again.
It’s not the life I would have chosen for him of course but it’s our routine now. I enjoy having a chat with our lovely nurses and getting jobs done round the house. Pudding gets lots more TV than usual and sometimes extra snacks as well. And I am so grateful for this man-made enzyme pumping round his body and unravelling some of the effects of Hunter Syndrome.