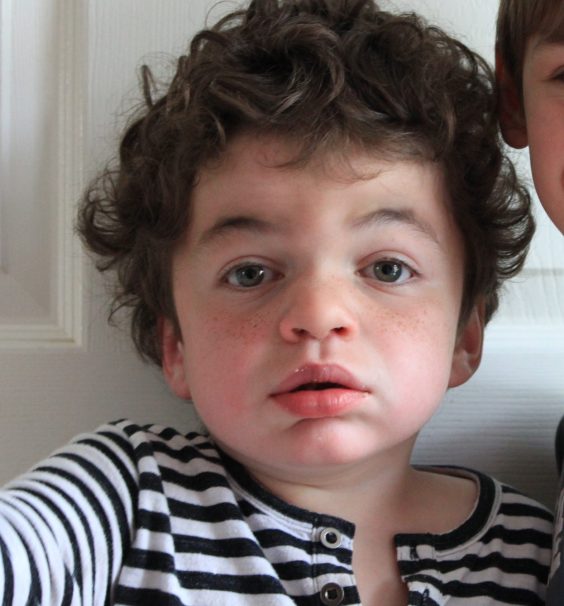
*Trigger warning – death and loss*
I’ve never really been a big fan of New Year’s resolutions. Why try and set yourself up to fail in the most miserable dark time of the year? (Eternal optimist, me!) But maybe this post does fall into the ‘New Year, new you’ sort of vibe, though the subject matter not so much.
When I set up this blog I promised myself and my readers that I would always be honest about what we’re going through and how I’m feeling. I didn’t think it would be fair to hide any aspect of our journey because the whole point of it is to share, so that hopefully others on the same path could recognise their own problems and feel a little less alone.
On the whole I’ve stuck with that, but there’s one area that I’ve often skirted around and avoided tackling head-on. Mostly because I was scared about how people would judge me after reading it. I’m still scared, but a conversation I had recently made me realise that I may not be the only person who has had similar thoughts on this topic.
I think about death quite a lot. You tend to when your child has been diagnosed with a life-limiting condition. (I never quite understood the family member that told another MPS parent that they ‘focus on death too much’.)
But of course death is never a simple subject to touch on. I’ve never lost anyone very close to me. Grandparents and friends, yes. But not a parent, or partner, or child. So I can only imagine the pit of grief that swallows you up after it happens. I know it can never be easy losing a loved one, whether it is fast or slow, expected or not. And I hope I will not offend anyone by what I am writing here. But it feels particularly cruel to face a long, slow decline for someone you love more than anything.
So here goes. Deep breath.
I have sometimes wished my son would die.
Not now. Not while he loves life and embraces it with such obvious relish. But if I could choose, I would choose a swift and merciful end for him rather than losing him bit by bit. And in my darker times I have wished that it was sooner rather than later, just to take away the agony of waiting for it.
One of the very first things I ever read about MPS when Pudding was diagnosed was the Wikipedia page. It refers to a case from 2004 when a father suffocated his 10-year-old son who had Hunter Syndrome. That has haunted me ever since. Never in a million years would I do something like that to my darling boy and this is not a blog post about mercy killing or euthanasia*, but I guess I understand part of why he may have done it.
Faced with the prospect of my son spending years losing the ability to talk (which he mostly has done), to walk, to swallow; thinking of him having more pain as his joints degenerate; knowing that he may be hit by seizures and by the end may not even recognise his family… there have been times where I’m certain I could not go through that. Maybe that makes me selfish, putting my wishes foremost.
Or is it? Is it selfish to hope that your loved one, whether 7 or 70, can live without pain, physical or mental, and can die with dignity? The reason we have these thoughts is because we love them. And love makes us want to end any suffering.
As with anything I write in this blog these are my own thoughts and emotions and I’m simply offloading. I may be in a tiny minority but I’m going to feel better for having said it. My thoughts may change, as so many things I’ve written about have done. Maybe as his condition worsens I’ll be more and more desperate for him to cling onto life with both hands and never let go.
Right now, as I’m writing this, Pudding is clambering onto my lap with his tablet, resting his head heavy against my cheek, his warm bulk blocking my view of the screen and making it pretty awkward to type. I simply can’t imagine him not being here.
But I will always hope for the best for him. In life and in death.

*I do happen to believe that an adult in their right mind with a life-limiting condition should have the choice to die at a time of their choosing, but know that this is a topic fraught with problems and presents a number of ethical issues.


 The lovely audiologist in the other room had the difficult task of trying to work out whether his reactions were genuine or whether he was anticipating the stimulus. The results agreed with the last hearing test he had, showing moderate hearing loss. But she wasn’t prepared to just accept that. She wants to be sure it’s a genuine result rather than just the difficulty of testing someone who doesn’t understand why we’re getting him to do this. So we’re going to try again another time, and also have someone observing him in school to see what he is like in a functional situation.
The lovely audiologist in the other room had the difficult task of trying to work out whether his reactions were genuine or whether he was anticipating the stimulus. The results agreed with the last hearing test he had, showing moderate hearing loss. But she wasn’t prepared to just accept that. She wants to be sure it’s a genuine result rather than just the difficulty of testing someone who doesn’t understand why we’re getting him to do this. So we’re going to try again another time, and also have someone observing him in school to see what he is like in a functional situation.
 I hate that the few other families who know and understand this MPS life are spread all over the world and often out of reach.
I hate that the few other families who know and understand this MPS life are spread all over the world and often out of reach.